
Complement deficiencies
Individuals with a complement deficiency can have clinical problems that are a result of the role that the specific complement protein plays in the normal function of the human body.
The more you understand about primary immunodeficiency (PI), the better you can live with the disease or support others in your life with PI. Learn more about PI, including the various diagnoses and treatment options.

Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.

Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.

Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.

Individuals with a complement deficiency can have clinical problems that are a result of the role that the specific complement protein plays in the normal function of the human body.
Complement activation is designed to eliminate invading microbes while producing minimal collateral damage. Complement proteins in the circulation are generally not activated until triggered by an encounter with a bacterial cell, a virus, an immune complex (antibody attached to a foreign material), or damaged tissue.
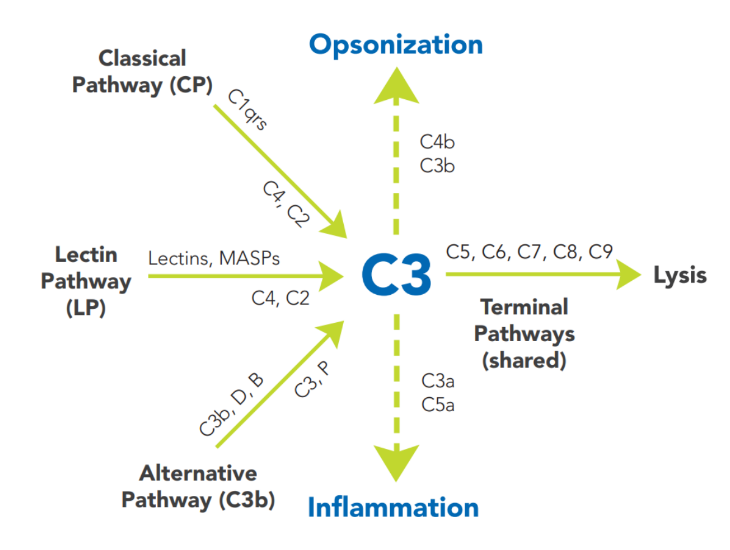
Complement activation is a cascading event with tremendous amplification potential. It must follow a specific order if the result is to be achieved. The circulating proteins have been grouped into three major activation pathways, based on the types of substances and proteins that initiate the activation. If you visualize a trident, the three tines (teeth) represent the different initiation routes of the complement system, while the handle represents the mechanism by which this cascade ultimately destroys the threat, no matter which activation pathway started the response. The diagram in Figure 22:1 depicts the activation pathways.
See if you qualify to participate in clinical trials evaluating new treatments and/or diagnostics for complement deficiencies.
Any complement deficiency should be treated as a form of PI, and the individual should be immunized against the bacteria that are most likely to infect them (see above). For example, boys with properdin deficiency should be immunized against Neisseria meningitidis, and meningococcal vaccines in addition to the usual childhood vaccinations. People with deficiencies of the other alternative pathway components and the terminal pathway proteins are also susceptible to Neisseria meningitidis and should be immunized. Antibody responses should be checked after vaccination, since the inability to activate complement may impair response to the vaccine.
Currently, there is no single treatment for complement deficiencies. Appropriate prevention and treatment of infections (usually with antibiotics) is key. Fresh frozen plasma infusions have been tried in some cases but carry a risk that the individual may make antibodies to the missing complement component, so prolonged use is not advised. In the case of HAE, effective medications to treat or prevent angioedema episodes are available. In aHUS or PNH, eculizumab, a complement-inhibitory antibody, may help. Prophylactic antibiotics can be used if the individual experiences repeated infections. Most of these individuals who are predisposed to infections eventually make antibodies against the offending bacteria and do not get sick as often.
The inherited deficiency of various complement proteins results in diseases due to the inability to perform primary immune functions, such as protection from infection or clearance of cellular debris. Additionally, in most situations where autoantibodies are present (such as SLE), these antibodies can engage complement, consume it, and result in tissue damage. Thus, there are certain acquired deficiencies of complement that are not inherited but can lead to disease. Both children and adults can be affected by complement deficiencies, so it is important to recognize the typical presenting symptoms of specific deficiencies as outlined above. Knowledge of the role of each complement component in the body and how it is regulated can help in understanding the effect of the specific deficiency and its treatment.
This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice.
Adapted from the IDF Patient & Family Handbook for Primary Immunodeficiency Diseases, Sixth Edition.
Copyright ©2019 by Immune Deficiency Foundation, USA
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.

