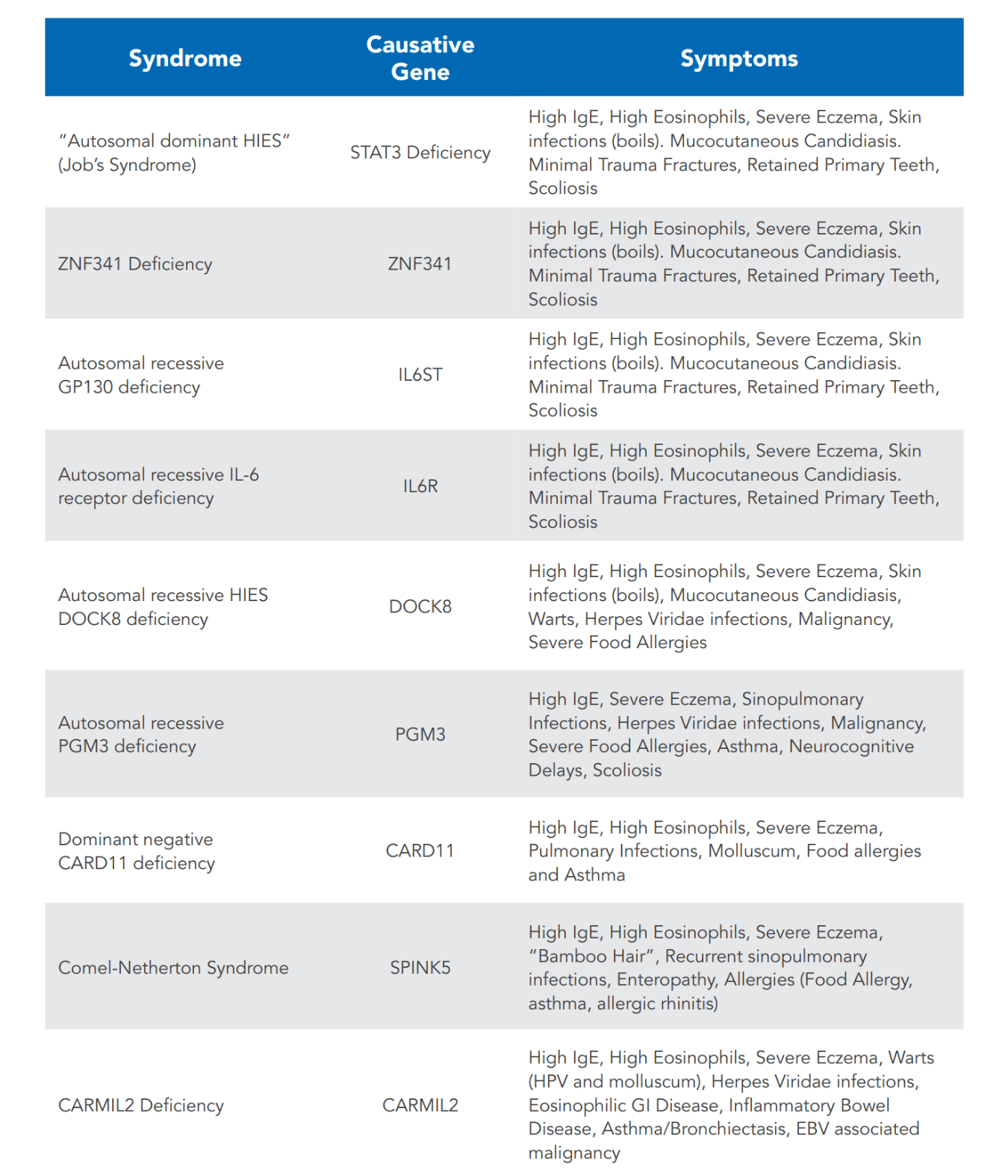
STAT3 deficiency is associated with heterozygous loss of function variants in the transcription factor STAT3. This is the more common form of HIES in the U.S. It commonly presents with skin findings, including newborn rash, eczema, and recurrent staphylococcal skin abscesses, as well as ear, sinus, and lung infections. The lung infections often result in cavitary lesions in the lungs (pneumatoceles).
Other common infections in STAT3 deficiency include mucocutaneous candidiasis (Candida fungus on mucous membranes and/or skin), manifesting typically as thrush, vaginal candidiasis, or candidal nail infection (onychomycosis), and recurrent shingles outbreaks. Additional findings include connective tissue and skeletal abnormalities, such as a typical facial appearance, hyper-extensibility of joints, retained primary teeth, recurrent bone fractures with minimal trauma, and a propensity to aneurysm formation of brain and cardiac blood vessels. Severe allergic diseases, such as food allergies, are not common in individuals with STAT3 deficiency. However, they often have positive skin prick tests making it appear that they have significant allergies.
A newborn rash or eczema is frequently the first manifestation of STAT3 deficiency. Pustular and eczema-like rashes usually begin within the first month of life, first affecting the face and scalp. Skin abscesses are a classic finding in this disorder, caused by a particular susceptibility to infections with Staphylococcus aureus. The degree of inflammatory symptoms, such as tenderness and warmth, often is quite variable. The term 'cold abscesses' is applied to those lesions that lack external signs of inflammation despite the presence of pus. The occurrence and severity of these abscesses substantially decrease with prophylactic therapy with antibiotics against Staphylococcus aureus. Deep tissue abscesses, such as liver or bone infections, are much less common in AD-HIES.
Recurrent bacterial cases of pneumonia are often encountered in individuals with AD-HIES. Pneumonias typically start in childhood and are most frequently caused by Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae. Similar to the occurrence of cold skin abscesses, these types of pneumonia may present with fewer symptoms than would be expected in a person with intact immunity. This relative lack of symptoms and subsequent delay in clinical presentation may contribute to advanced disease and significant tissue damage before identification and initiation of appropriate therapy.
Infection-induced tissue destruction in individuals with AD-HIES may give rise to pneumatocele formation, and large cavities in the lung, which is a distinguishing feature of AD-HIES due to STAT3 deficiency. When pneumatoceles or bronchiectasis are present in the lungs, the individuals are more susceptible to lung infections with other bacteria, such as Pseudomonas, and molds, such as Aspergillus.
The involvement of both the connective and skeletal tissues is an important feature of STAT3 deficiency. An asymmetrical facial appearance with a prominent forehead and chin, deep-set eyes, broad nose, thickened facial skin, and a high-arched palate are typical of this disorder. These features evolve during childhood and become more established by adolescence.
Individuals with AD-HIES also exhibit hyperextensibility of the joints. They frequently suffer bone fractures from seemingly insignificant trauma and bone density may be reduced. Scoliosis is common and typically emerges during adolescence or later in life. Craniosynostosis (fused skull bones) and extra or abnormally formed ribs or vertebrae are also found more often in those with AD-HIES than in the general population. These skeletal abnormalities are not usually seen in individuals with DOCK8 deficiency.
Abnormalities affecting the teeth are another common feature of AD-HIES with STAT3 mutations. Retention of primary teeth, even after the permanent teeth have erupted, is a consistent finding. This abnormality is revealed on panoramic x-ray views as double rows of retained primary teeth overlaying the permanent ones. Surgical extraction of the retained primary teeth is often necessary for healthy dentition. Children who have had their retained primary teeth extracted will have a normal eruption of their permanent teeth.