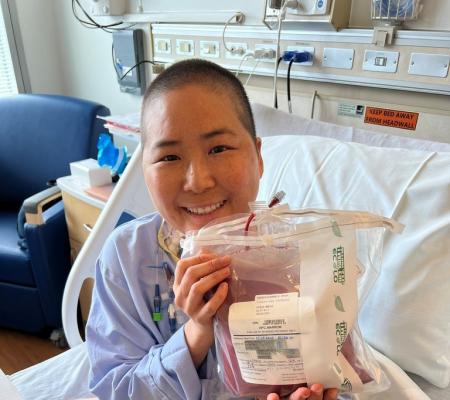
Treatment
The treatment of CVID is similar to that of other disorders with low levels of serum immunoglobulins. In the absence of a significant T lymphocyte defect or organ damage, Ig replacement therapy almost always brings improvement in symptoms. Immunoglobulin is extracted from a large pool of human plasma; it consists mostly of IgG and contains all the important antibodies present in the normal population.
People with chronic sinusitis or chronic lung disease may also require long-term treatment with broadspectrum antibiotics. If mycoplasma or other chronic infections are suspected, antibiotics specific for those organisms may be indicated. If bronchiectasis has developed, a daily pulmonary regimen (chest physiotherapy and postural drainage) may be needed to mobilize the secretions from the lungs and bronchi and make them easier to cough up.
When individuals with CVID are on Ig replacement therapy, routine immunizations are not required because the Ig solutions contain protective antibodies against these. Exceptions may be the new killed shingles vaccine, the human papillomavirus vaccine (HPV), and the annual viral influenza vaccine.
Those with GI symptoms and malabsorption should be evaluated for the presence of Giardia lamblia, rotavirus, Campylobacter, norovirus, bacterial overgrowth, and other GI infections. In some cases, inflammatory bowel disease is found. This is an autoimmune condition that can be treated by the medications normally prescribed for individuals who are not immunodeficient, including the newer biologic drugs. Maintaining a balance between the immunosuppression used to control the autoimmune process while avoiding compounding the defects of the underlying PI requires close cooperation between the individual and the various specialists involved in their care.
If autoimmune or inflammatory disease, granulomas, or tumors develop, the treatment is usually the same as would be given to a person with a normal immune system. When people with CVID, however, have these complications, there is a tendency for them to be less responsive to therapy. Regular checkups, including lung function, are recommended.
Most individuals with CVID carry out most, if not all, normal activities. Regularly scheduled and careful follow-up is still mandatory as new problems may arise or evolve over time. In general, those who are stable are seen at least yearly, but with added questions or when other conditions arise, shorter intervals, such as three to six months, are needed. In addition to addressing routine questions and checking blood counts, metabolic panels, and Ig levels, monitoring for weight changes is important, as Ig doses may need to be adjusted. There is no current consensus on how best to monitor for lung disease. Chest X-rays are not able to show the same level of detail as a CT scan, but there is a lower radiation exposure with a chest x-ray. For more frequent follow-up of those with chronic cough and/or known lung damage, complete lung functions, including carbon monoxide (CO) diffusion, is useful, with possible chest CT at 3-4 year intervals. Monitoring for autoimmunity is usually accomplished through routine blood counts and general medical oversight, which will reveal characteristic symptoms. GI diseases will be similarly evident with complaints of diarrhea and, often, weight loss. Routine endoscopy is not required although people with suggestive GI symptoms should have appropriate upper and/or lower endoscopy (colonoscopy) with examination for H pylori, pathogenic bacteria or viruses, or other mucosal changes. Loss of height may reflect loss of bone density; this requires attention to vitamin D, calcium, and other standard therapies. Evaluation of enlarged lymph nodes is not simple. When new lymph nodes appear and persist, biopsy may be required, but most commonly, these reflect simply reactive changes that are not clinically significant.