
DiGeorge or 22q11.2 deletion syndrome
DiGeorge syndrome, most frequently caused by a deletion at 22q11.2, is a PI caused by abnormal migration and development of certain cells and tissues during fetal development.
The more you understand about primary immunodeficiency (PI), the better you can manage it. Learn about PI diagnoses and treatment options.

Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.

Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.

Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.

DiGeorge syndrome, most frequently caused by a deletion at 22q11.2, is a PI caused by abnormal migration and development of certain cells and tissues during fetal development.
See if you qualify to participate in clinical trials evaluating new treatments and/or diagnostics for DiGeorge syndrome or 22q11.2 deletion syndrome.
The treatment of a child with DGS varies depending on the infant’s immune status and the medical problems that the child has. For the 1% of infants with DGS who, on newborn screening, are found to have no T cells, a transplant of the thymus is recommended because of the high risk of infection and death. Because the defect in T cell development is due to a lack of thymus, hematopoietic stem cell transplantation (bone marrow transplantation) is not effective. These infants should be kept in strict isolation to avoid infection until T cells develop after thymus transplantation.
For all infants with DGS, a multidisciplinary team is the preferred form of care. One example of such a team would be a clinic specializing in 22q11.2DS with multiple medical specialists needed for any child with DGS (whether or not the child has 22q11.2DS). The table below lists specialists who may be needed to care for infants with DGS. When a team is not available, the pediatrician, geneticist, or immunologist caring for the child should be asked to arrange for the appropriate doctors to be consulted for care for the child.
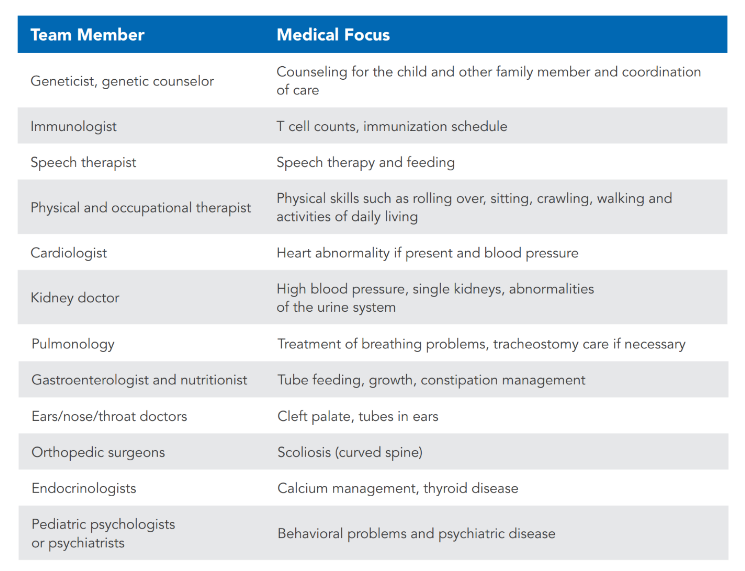
For the 99% of infants with DGS who have T cells, the immune system is not a major problem. An immunologist will determine if the T cells are high enough for administration of live vaccines such as the rotavirus vaccine; the measles, mumps, rubella virus vaccine; and the varicella (chickenpox) vaccine. An immunologist should assess each child periodically to confirm that the T cell numbers remain adequate.
T cells help another white blood cell called the B cell make antibody responses to vaccines. In about 10% of individuals with 22q11.2 DS, they have difficulty with production of immunoglobulins or vaccine responses. Some of these individuals require immunoglobulin replacement therapy.
There is great variability in the other problems that a child with 22q11.2 DS may have.
Some children with very mild forms of DGS are diagnosed later in life due to speech abnormalities or other subtle findings, while others have varying degrees of impairment in any combination of the following aspects of DGS.
Heart defects at birth often need to be repaired by heart surgery. Cardiac follow-up will be needed depending on the type of heart defect. The cardiologist may also provide guidance for high blood pressure (if present).
Low calcium levels from a poorly functioning parathyroid gland usually improve, and most children do not need calcium replacement for more than one year. The exception is the group born with no T cells—these children often remain on calcium for life. It is important that calcium levels be followed by the pediatrician because on rare occasions the calcium levels may drop in a child who previously had been able to come off of calcium replacement.
The thyroid should be checked annually in children with DGS because thyroid disease is very common. By the time an individual with DGS is 40 years old, 1 of 5 have thyroid disease and are treated with thyroid medication.
Learning disabilities are common. Most children with DGS take longer than usual to learn to talk. Math is particularly difficult. Working with school officials, including the school nurse, to develop an Individualized Education Program (IEP) and/or an Individual Healthcare Plan (IHP) can be very helpful.
Psychiatric disease can develop in adolescents and adults. Two common conditions are schizophrenia and bipolar disorder in individuals with 22q11.2DS. Family and school support are very important for children with DGS to enhance their development as best as possible. Staying connected to others through the Immune Deficiency Foundation, the 22q11.2 Society, and the CHARGE Syndrome Foundation can be helpful for families.
Rethymic
Allogeneic processed thymus tissue–agdc.
Approved to treat: Congenital athymia.
Report side effects/adverse reactions at 833-369-9868.
The CHARGE Syndrome Foundation champions the lifelong potential of people with CHARGE syndrome through outreach, education, and research. The CHARGE Syndrome Foundation was founded in 1982 in Columbia, Missouri. Today, the organization offers a biennial international conference, support for clinical and scientific research, and support and resources for families.
The International 22q11.2 Foundation is a nonprofit organization dedicated to supporting the needs of families and individuals affected by chromosome 22q11.2 differences by promoting awareness, state-of-the-art clinical care, cutting edge research endeavors, and solidarity with related associations around the globe.
This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice. Additionally, links to other resources and websites are shared for informational purposes only and should not be considered an endorsement by the Immune Deficiency Foundation.
Adapted from the IDF Patient & Family Handbook for Primary Immunodeficiency Diseases, Sixth Edition.
Copyright ©2019 by Immune Deficiency Foundation, USA
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.



