
Why laboratory-developed tests’ fate matters for rare disease patients
June 24, 2022
The more you understand about primary immunodeficiency (PI), the better you can manage it. Learn about PI diagnoses and treatment options.

Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.

Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.

Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.
For decades, there have been two types of patient sample tests in the U.S.—commercialized diagnostic tests and what are called “laboratory-developed tests” (LDTs). Unfortunately, proposed legislation called the Verifying Accurate Leading-edge IVCT Development Act (VALID Act), which is designed to streamline regulations and provide greater oversight for both types of tests, could have the unintended consequence of putting up roadblocks for those with rare diseases.
IDF is closely monitoring the VALID Act and its potential implications for the primary immunodeficiency (PI) community.
While a commercial test and an LDT might very well measure the exact same thing using the exact same method, differences in how they are developed and used ultimately spell out different regulatory paths.
Commercial diagnostic tests are typically developed by for-profit companies and sold as kits to clinical laboratories nationwide. For example, your doctor’s office might send your blood sample to a lab that uses a commercial kit to test your immunoglobulin levels.
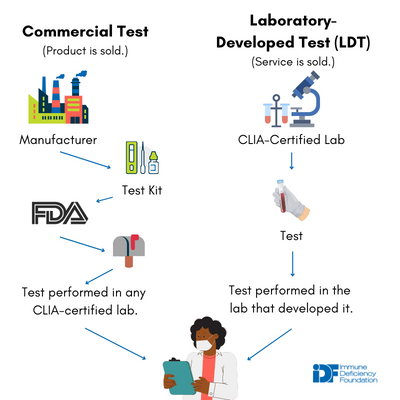
Like medical treatments, commercial diagnostic tests are regulated by the U.S. Food and Drug Administration (FDA) and require a rigorous pre-market review process to establish that the kits work, are reliable, and don’t cause harm to patients. Because of the economics of commercial test development and regulation, rare diseases typically do not have commercialized tests available on the market—the cost is simply too high to justify selling a test specific to a very small group of people.
LDTs, in contrast, are tests that are developed and used in only one laboratory. Samples can be sent to that laboratory to be analyzed using the LDT, but the LDT itself cannot be sold to other laboratories. LDTs are frequently developed to meet a clinical need, for example, the diagnosis of a rare condition that does not have a commercial test available. While for-profit companies do develop LDTs, they are also widely developed by academic, hospital-associated, and government public health laboratories.
Unlike commercial tests, LDTs are not regulated by the FDA. Instead, they are regulated under the Clinical Laboratory Improvement Amendments (CLIA) by the Center for Medicare & Medicaid Services (CMS). Under CLIA, the laboratories that develop LDTs undergo rigorous certification processes, must hire personnel with particular qualifications, and must keep data on the analytical validity (i.e., does the test accurately measures what it is supposed to measure?) of all LDTs performed in the lab up-to-date and accessible to CMS. The CLIA model regulates the laboratory developing and performing the LDT more heavily than the LDT itself.
Given that LDTs are often developed for unmet clinical needs, such as the diagnosis and progression of rare diseases, they are crucial to the primary immunodeficiency community. In fact, the rapid growth in the number of recognized, distinct PIs over the last 20 years is the direct result of cutting-edge LDTs, often based on sequencing patients’ genomes for rare genetic variants.
LDTs are also crucial for newborn screening conducted in state public health laboratories. Because screening is performed on every newborn (not just those with suspected conditions), commercial newborn screening tests are economically viable and are available for many conditions.
However, commercial kits typically require laboratories to purchase and use the manufacturer’s proprietary testing machinery. Instead of purchasing multiple instruments, many state laboratories have opted to develop LDTs that work on the instruments they already have. In fact, IDF found that 90% of state laboratories screening for severe combined immunodeficiency (SCID) use LDTs even though commercial tests are available. Although SCID is currently the only PI included in newborn screening, other PIs are likely to be included in the future.
The bottom line is, for newborn screening, LDTs are more economical and are easier to integrate into a state laboratory’s existing setup and processes.
The disparity in regulations between commercial diagnostic tests and LDTs has been raised as a loophole in protecting patients from fraudulent or meaningless tests. In fact, several companies, including Elizabeth Holmes’ Theranos, have been able to market unproven tests by offering them as LDTs. Non-commercial labs typically publish scientific papers on how their LDTs work, but because commercial labs consider that information proprietary, their LDTs are often 'black box' tests.
Because of these issues, clinical testing experts agree that LDTs need more oversight. How to provide that oversight is an open question, however.
The VALID Act is the most recent legislation attempting to increase oversight of LDTs. The VALID Act would create a new FDA category called in vitro clinical tests (IVCTs) that merges commercial diagnostic tests with LDTs. IVCTs would undergo a regulatory process similar to the current process for commercial diagnostic tests, including registration with the FDA and, in some cases, pre-market review. FDA pre-market review looks at a test’s analytical validity as well as its clinical validity (i.e., is the measurement useful in diagnosis or treatment decisions?).
Meanwhile, CLIA regulations for LDTs still stand. If the VALID Act passes, laboratories that perform LDTs will be regulated twice over by two different agencies. The burden is not just in paperwork. Both CLIA and FDA are funded through user fees (which means the ‘users’ of their services pay to fund each agency’s operations), so laboratories performing LDTs will have to pay for both CLIA and FDA regulations.
For academic, hospital-associated, and state public health laboratories, these burdens may put them out of the LDT business, even if the LDTs they offer are largely exempt from FDA’s pre-market review.
Where does that leave patients? The VALID Act may have the unintended consequence of shifting LDTs to only well-resourced, for-profit labs. What happens if those labs decide that LDTs for rare conditions are not profitable enough, as their counterparts in commercial test development have? Will rare disease diagnostic and progression testing go the way of rare disease treatments, where treatments that work still struggle to make it to patients?
Stay tuned as IDF continues to monitor the situation. In the meantime, read the IDF Medical Advisory Committee’s letter to the Senate HELP Committee on the VALID Act.
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.



