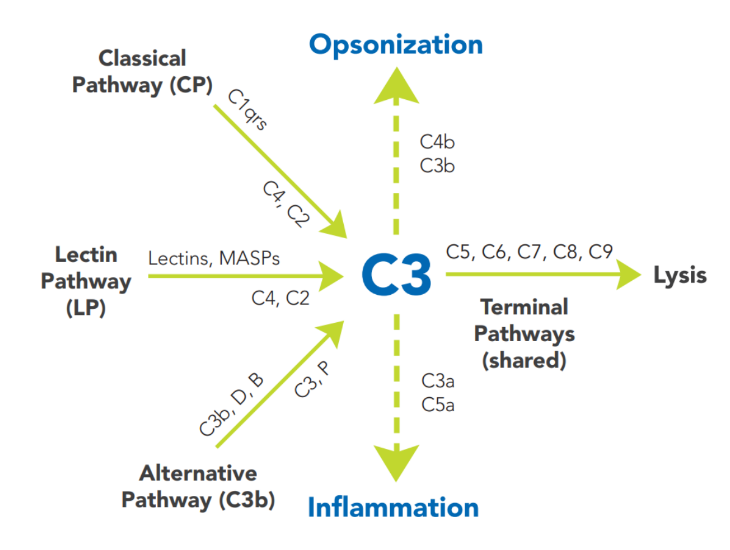
The terminal pathway (TP) is the final set of steps in the complement activation process that forms a membrane lesion or hole (membrane attack complex, MAC), thus killing susceptible pathogens (bacteria) or other cells that activate complement on their surfaces. The TP is dependent upon at least one of the other pathways, such as AP, CP, or LP for activation. The components of the TP are C5, C6, C7, C8 and C9, and they form the MAC (C5bC6C7C8C9, or C5b-9).
Regulatory proteins to prevent unregulated activity (and tissue damage) are present as control mechanisms for each pathway. These can be present in the fluid phase, such as blood, or on the cell surface. C1-esterase inhibitor (C1-INH) is a serine protease inhibitor (SERPIN) in the blood and acts by forming a complex with active enzymes to trap and inactivate them. It is important in controlling the C1r and C1s activation in the classical pathway and the MBL-associated serine proteases in the lectin pathway along with enzymes in the coagulation system. Factor H and Factor I are also fluid phase proteins that help to regulate the amplification of the complement system. Their deficiency can result in a condition known as atypical hemolytic uremic syndrome (aHUS), which is associated with red blood cell damage and kidney dysfunction. CD46 and CD55 are proteins present on the cell surface, that also limit complement activation on the cell from which they are expressed. CD59 deficiency is associated with lysis of red blood cells and kidney damage, as seen in paroxysmal nocturnal hemoglobinuria (PNH).
The dynamic interplay among the different complement pathways and their control processes involves other plasma protein systems such as enzymes of the coagulation system, enzymes from inflammatory cells, and substances such as histamine and elastase released from cells in the local environment. All of these participants affect the outcome of an activation event. Most of the time, the outcome is favorable to the host. The diseases that accompany uncontrolled activation or a defect in the function of complement are often the result of an inherited deficiency or subtle impairment of one or more of its regulators.