
-
Understanding primary immunodeficiency (PI)
Understanding PI
The more you understand about primary immunodeficiency (PI), the better you can manage it. Learn about PI diagnoses and treatment options.
-
Living with PI

Living with PI
Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.
-
Get involved

Get involved
Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.
-
Advancing research and clinical care

Advancing research and clinical care
Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), these resources will help you advance the field. Get details on surveys, grants, and clinical trials.

Key points:
- Gene therapy for primary immunodeficiency (PI) modifies hematopoietic stem cells from the bone marrow to restore a working immune system.
- Currently, gene therapies for Wiskott-Aldrich syndrome (WAS) and leukocyte adhesion deficiency type I (LAD1) are the only FDA-approved gene therapies available in the U.S. for PI. Other gene therapies are available through clinical trials only.
- There are two main strategies for PI gene therapy: gene addition, which inserts a working gene copy into cells, and gene editing, which directly corrects PI-causing genetic variants. Both methods use viral vectors to deliver the therapy to cells.
- Gene therapy has the potential to treat many types of PI and it is likely that gene therapy will be the standard treatment for severe PI in the future.
The goal of gene therapy is to provide a long-term treatment for genetic conditions by modifying the DNA in a person’s cells. In the case of primary immunodeficiency (PI), gene therapy focuses on hematopoietic stem cells in the bone marrow, which make all of a person’s immune system cells. By altering these stem cells, gene therapy can provide people with PI with a working immune system.
Currently, gene therapy for primary immunodeficiencies (PIs) is mainly available through clinical trials in the United States. There are two exceptions:
- Etuvetidigene autotemcel (trade name Waskyra), which is approved by the U.S. Food and Drug Administration (FDA) to treat Wiskott-Aldrich syndrome (WAS) [1].
- Marnetegragene autotemcel (trade name Kresladi), which is approved to treat leukocyte adhesion deficiency type I (LAD1).
However, several research groups are actively working toward FDA approval, and there may be FDA-approved gene therapies for additional types of PI in the near future. Additionally, the European Medicines Agency (EMA) authorized Strimvelis, a gene therapy for a type of severe combined immune deficiency caused by adenosine deaminase deficiency (ADA-SCID), in Europe [2].
How gene therapy works
Strategies for correcting cells
Researchers have developed several approaches for gene therapy. The two main strategies used for treating PI are gene addition and gene editing.
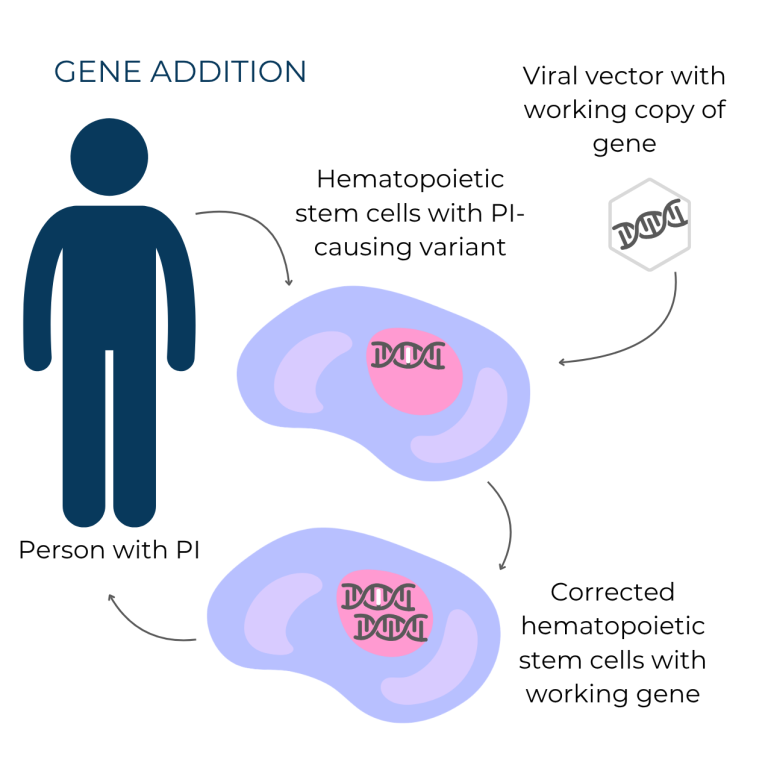
Gene addition involves adding a working copy of a gene to cells and was the first type of gene therapy researchers developed. Gene addition is typically used to treat autosomal recessive or X-linked forms of PI. Once inside cells, the working gene copy becomes part of the cells’ DNA, and it is passed on to all future cells produced by the corrected cell. In the case of PI, this means that all blood and immune system cells made by the modified hematopoietic stem cells carry the extra working copy of the gene. Waskyra for WAS and Kresladi for LAD1 both use the gene addition strategy.
In contrast, gene editing directly targets and repairs the genetic variant causing the condition. This type of gene therapy is generally more precise and can treat a broader range of genetic variants. Once the pieces of the gene editing system are delivered to cells, they locate the PI-causing gene variant in the DNA and correct it, restoring the gene’s function. As with gene addition, the correction is permanent and all future cells produced by the corrected cell carry the edited, working version of the gene.
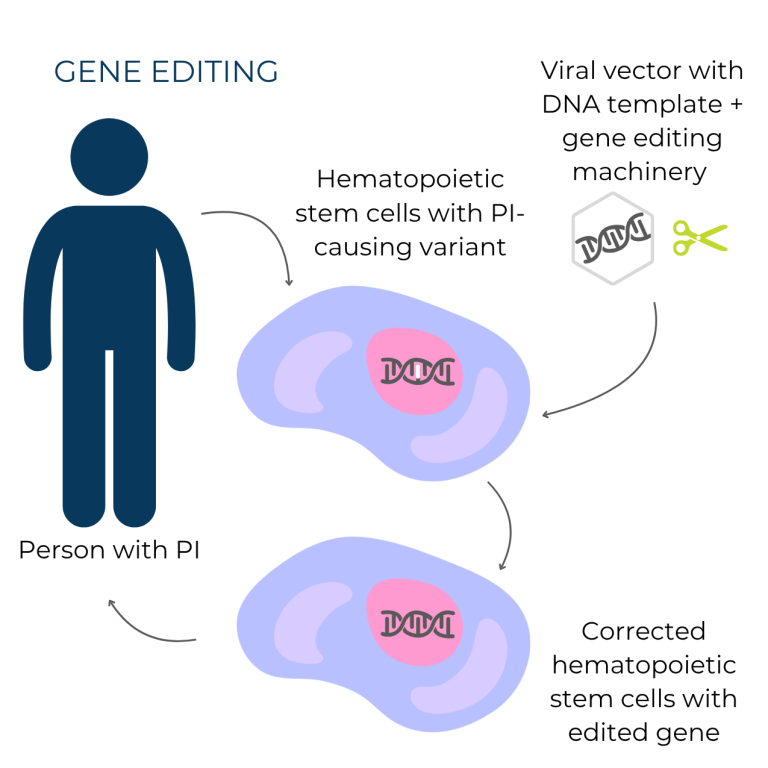
For both gene addition and gene editing, the therapy has to get inside of cells. Most gene therapies use viral vectors—virus shells that cannot cause infections—to get the therapy into the cells. Viral vectors can’t cause infections because the virus' genes are replaced by the gene therapy cargo, which is the working gene copy that will be added into an individual’s DNA (gene addition) or used to correct a disease-causing variant (gene editing).
To date, most FDA-approved gene therapies for non-PI conditions use gene addition rather than gene editing. However, in 2023, FDA approved the first gene editing therapy for treating sickle cell disease [3]. Gene editing is also being tested in clinical trials for several conditions, including hereditary angioedema (HAE), which is a deficiency of C1 inhibitor [4], and chronic granulomatous disease (CGD) [5]. Gene editing technology continues to advance, offering potential treatments for a broader range of genetic disorders.
Receiving gene therapy
Gene therapy is either ex vivo (outside the body) or in vivo (inside the body), referring to the location of the person’s cells when they are treated.
Waskyra for WAS and Kresladi for LAD1 are both ex vivo gene therapies. For ex vivo gene therapy, the person with PI serves as both the donor and recipient in a process similar to hematopoietic stem cell transplantation (HSCT). Doctors first collect hematopoietic stem cells from the individual's bone marrow, peripheral blood, or umbilical cord blood (in the case of a newborn). The laboratory then grows the cells, typically with proteins called growth factors that tell the cells to grow and divide. During this time, the cells are modified either by gene addition using a viral vector or gene editing using a system like CRISPR/Cas9 combined with a viral vector. Once the cells are modified, they are typically infused into a large vein in the same way that someone receives a blood transfusion or stored for later use.
Before the modified hematopoietic stem cells can be infused, the individual often receives conditioning with chemotherapy or immunosuppressive drugs. This conditioning, usually at a lower dose than with HSCT, helps create space in the bone marrow for the corrected stem cells to grow. Since ex vivo gene therapy uses the individual’s own stem cells, there is no risk of graft rejection or graft-versus-host disease (GVHD), which can occur with HSCT. After conditioning, the corrected stem cells are infused through an IV line, where they travel to the bone marrow and begin producing a healthy immune system. Healthcare providers monitor the individual closely for signs of new immune cells or infection during recovery in a hospital setting.
In vivo gene therapy treats individuals without removing cells from the body. The key advantage is that in vivo gene therapy eliminates the need for cell collection, which could expose the cells to factors that interfere with their growth and overall health. However, a major challenge of in vivo gene therapy is making sure that the gene therapy cargo reaches the correct cells, while minimizing effects on other cells. Currently, in vivo therapies often involve direct injections into specific areas, such as the eye, which is a self-contained environment that helps minimize the risk of impacting other cells.
History of gene therapy to treat PI
Gene therapy has been used in clinical trials to treat people with several types of severe combined immunodeficiency (SCID), hereditary angioedema (HAE) type 1, X-linked chronic granulomatous disease (X-linked CGD), and leukocyte adhesion deficiency-1 (LAD-1). In 2025, Waskyra for WAS became the first gene therapy for PI approved in the U.S.
Gene therapy for ADA-SCID
The first clinical trial for gene therapy was at the National Institutes of Health in 1990 and treated a 4-year-old girl with ADA-SCID [6]. The design of this first trial focused on adding the ADA gene sequence to her T cells with a viral vector. She received multiple infusions of her own gene-modified T cells over a two-year period. Her T cell counts and function rapidly rose to normal levels after treatment. Although her total number of T cells eventually fell below the range for healthy individuals after about 9 years, she remained healthy and free from serious infections. The working ADA gene remained in 20-40% of the patient’s T cells at last report, twelve years after gene therapy [7].
After this initial clinical trial showed that gene therapy could be done safely and that gene-corrected T cells could survive and function for years, additional clinical trials began to treat other children with ADA-SCID by targeting the long-lasting hematopoietic stem cells from the bone marrow. In terms of providing a working immune system, the results have been excellent. Most of the nearly 100 individuals treated for ADA-SCID reached a significant, long-lasting increase in T and B cell counts and have had a remarkable improvement in immune function [8], [9].
In 2016, the European Medicines Agency (EMA) approved Strimvelis, a gene therapy for ADA-SCID that uses a retroviral vector. Thirty-three patients were treated with Strimvelis before reports in the fall of 2020 that one patient developed leukemia, a type of blood cancer, four years after treatment due to insertional mutagenesis [10]. Insertional mutagenesis is when DNA from the viral vector activates other genes near where the new, working gene has been added to the stem cell’s chromosome. Depending on which genes are nearby, activating these genes can make a stem cell more likely to become cancerous. Gene therapy research now uses other vectors that are less likely to cause insertional mutagenesis.
EMA issued a direct healthcare professional communication in early 2021 advising clinicians to follow patients closely for signs of leukemia [11]. Strimvelis remains authorized in the European Union.
Gene therapy for other PIs
The first gene therapy trials for X-linked SCID, X-linked CGD, and WAS also used retroviral vectors to add working versions of the affected genes into hematopoietic stem cells. While these trials resulted in restored immune function for the majority of treated individuals, a substantial number of people went on to develop leukemia in the years following treatment due to insertional mutagenesis caused by the viral vector.
Recent trials for several types of PI use updated viral vectors, called lentiviral vectors, that are designed to avoid insertional mutagenesis. These therapies resulted in immune benefits that are similar to, or even better than, those seen in earlier trials. To date, no cases of leukemia have been associated with lentiviral vector-based gene therapies for PI. However, there have been cases of insertional mutagenesis in a lentiviral vector trial for a metabolic disease called adrenoleukodystrophy [12]. This complication underscores the importance of ongoing monitoring and further advancements in the technology to ensure safety.
Waskyra for WAS and Kresladi for LAD1 use lentiviral vectors and both carry a warning that healthcare providers should monitor all treated individuals for cancer for at least 15 years after treatment [13].
Limitations of gene therapy
Gene therapy shares some limitations with HSCT. Like HSCT, gene therapy will fix problems specific to the cells made from the modified hematopoietic stem cells, but problems outside of the blood or immune system may not be corrected. As a result, PIs that affect cells outside the blood may not be fully treated. In addition, sperm and egg cells are also not modified, so a person who has had successful gene therapy can still pass PI-causing variants on to their children. Gene therapy also cannot reverse or repair damage already caused by PI, such as lung damage from severe infections.
Unlike HSCT, which is a general therapy that can treat a wide variety of PIs, gene therapy is more specialized and requires a tailored approach for each specific type of PI. This specialization makes gene therapies some of the most expensive treatments available. Health insurance companies may not cover them, even after FDA approval. Furthermore, gene therapy is limited to treating PIs caused by variants in a single, identified gene.
Despite these challenges, growing experience with gene therapy demonstrates its potential to successfully treat PI through gene addition or editing. As the field continues to develop, gene therapy may become a treatment option for many severe types of PI in the future.
Gene therapy products approved in the U.S.
Kresladi
(Manufacturer: Rocket Pharma
)
Marnetegragene autotemcel.
Approved to treat: Leukocyte adhesion deficiency type 1 in people without a matched, sibling donor suitable for hematopoietic stem cell transplant (HSCT).
Ages: N/A.
Waskyra
(Manufacturer: Fondazione Telethon ETS
)
Etuvetidigene autotemcel.
Approved to treat: Wiskott-Aldrich syndrome (WAS) in people without a matched, related donor suitable for hematopoietic stem cell transplant (HSCT).
Ages: 6+ months.
Latest gene therapy resources


This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice. Additionally, links to other resources and websites are shared for informational purposes only and should not be considered an endorsement by the Immune Deficiency Foundation.
Adapted from the Patient & Family Handbook for Primary Immunodeficiency Diseases, Sixth Edition.
Copyright ©2019 by Immune Deficiency Foundation, USA.
Sign up for updates
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.
