
-
Understanding primary immunodeficiency (PI)
Understanding PI
The more you understand about primary immunodeficiency (PI), the better you can manage it. Learn about PI diagnoses and treatment options.
-
Living with PI

Living with PI
Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.
-
Get involved

Get involved
Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.
-
Advancing research and clinical care

Advancing research and clinical care
Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.

Key points:
- Genes are segments of DNA that are organized into structures called chromosomes. Genes give your body instructions on how to grow, develop, and function.
- Primary immunodeficiencies are caused by specific “spelling changes” in certain genes that cause the gene to not work properly. These spelling changes are called variants and can be passed down from parents to children (inherited) or can be brand new in a child (de novo).
- There are many different ways, called inheritance patterns, that genetic conditions like PI can be passed down in families.
- Some variants that cause PI are not inherited or de novo. Instead, these somatic variants happen later during development and are found in only some, but not all, of a person’s cells.
Primary immunodeficiencies (PIs) are mostly caused by changes, or variants, in genes that are important for how the immune system works. Many of these genes have functions outside of the immune system too and, as a result, some PIs affect other biological processes like muscle function or brain development.
How human genetics works
Chromosomes and DNA
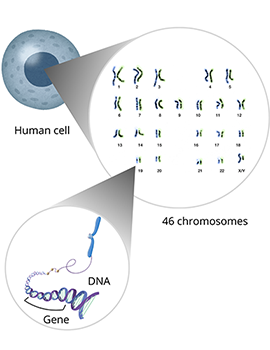
Cells are the building blocks of all living things. Humans have trillions of cells that work together to help our body grow and function. At the center of each cell in the human body are 46 individual pieces of deoxyribonucleic acid (DNA) called chromosomes. This set of chromosomes contains instructions called genes for building every protein, cell, and organ in your body, all coded within the DNA. Together, your chromosomes are like a set of recipe books containing all of the gene recipes necessary to make you.
To get 46 chromosomes total, each person gets two sets of 23 chromosomes, one set from each biological parent. During fertilization, the egg, which contains a set of chromosomes from your biological mother, comes together with the sperm, which contains a set of chromosomes from your biological father. This way, each parent contributes half of the genetic information for their child.
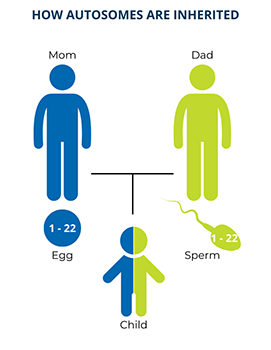
Twenty-two of the chromosomes in each set are numbered and these are called autosomes. Chromosomes of the same number in each set have the same genes on them. As a result, one copy of each autosomal gene comes from a person’s biological mother and the other copy comes from their biological father. This means that everyone, regardless of sex, has two copies of every autosomal gene.
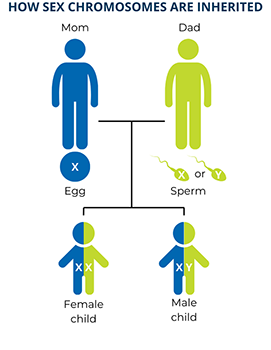
The last chromosome in each set is a sex chromosome. Humans have two types of sex chromosomes, X chromosomes and Y chromosomes, that have different sets of genes. A person who has two X chromosomes is genetically female and a person who has one X and one Y chromosome is genetically male. Since biological mothers can only contribute an X chromosome to their children, the sex of a child is determined by which sex chromosome (X or Y) the biological father contributes through his sperm. This also means that genetic females have two copies of each gene on the X chromosome, while genetic males have only one copy of each gene on the X chromosome, passed down from their mothers.
Inside each cell, there are also structures called mitochondria that generate and store energy. Mitochondria have their own DNA (mtDNA) with a few gene ‘recipes.’ mtDNA is only inherited from a person’s mother because only the egg contributes mitochondria to the developing baby. At this time, no mitochondrial genes have been associated with PI, but people who undergo genetic testing may receive a separate mtDNA report.
Genes and gene variants
There are four individual building blocks, also known as nucleotides, that make up DNA: adenine (A), thymine (T), guanine (G), and cytosine (C). The order of these nucleotides, or the DNA "sequence," is how the long chains of DNA store the information for making proteins and other molecules. This encoding is similar to how the order of letters from the alphabet form words, sentences, and paragraphs. A gene is the DNA sequence that codes for one specific protein or other molecule.
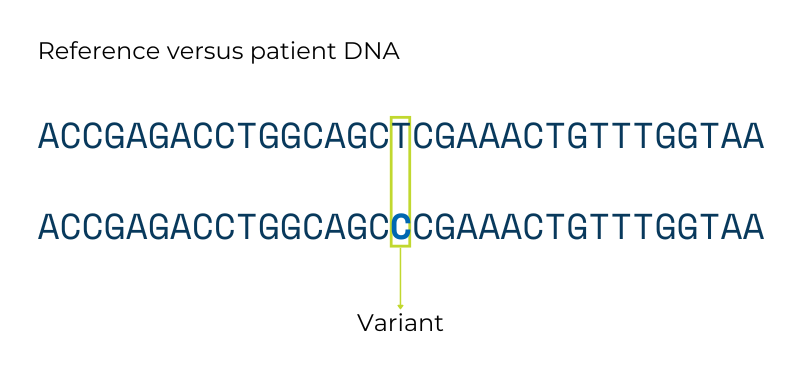
A gene variant is a difference in the DNA sequence of the same gene between people. Any two people who are not identical twins have many, many sequence differences between their genes. Some gene variants do not lead to a big change in the instructions they give the body and do not cause problems. These types of variants are like spelling the same word two different ways (e.g., the color “gray” vs “grey”).
Other gene variants do significantly change the instructions they give to the body and can cause genetic disorders like PI. These types of variants are also called mutations or pathogenic variants in the medical community. Because parents contribute genetic information to their children, parents with variants that cause PI can pass these down to their children. No one has control over their genetics or which variants are passed on to their children.
Sometimes, variants randomly happen when DNA is copied to make egg or sperm cells, or when a fertilized egg begins growing into a baby. These variants are brand new in a child and are not passed down from either parent. These are called de novo variants and de novo variants can also cause genetic conditions like PI. In these cases, neither of the parents have the variant, but the child does and can then pass the variant on to their children.
Parents cannot control what genetic variants arise in or are passed on to their children—genetic disorders are no one’s fault.
Ways that gene variants cause PI
Gene variants that are passed down from a child’s biological parents or that arise de novo cause three different types of PI: autosomal recessive, autosomal dominant, or X-linked disorders. These terms refer to how the disorder is passed down, or inherited. They apply to de novo variants as well because the person with the de novo variant can pass that variant on to their children even though they didn’t inherit it themselves.
Although treatments like hematopoietic stem cell transplantation (HSCT) or gene therapy may be curative, they do not change or replace a person’s egg or sperm cells. Because of this, someone successfully treated with HSCT or gene therapy can still pass on their PI-causing variant and can have a child with PI.
Autosomal means that the PI-causing variant is in one of the genes on an autosome, so both males and females have the same chance of inheriting the variant.
X-linked means that the PI-causing variant is in one of the genes on the X chromosome. Since the X chromosome is a sex chromosome, the chance of a child being affected by an X-linked disorder depends on the sex of the child. In the past, X-linked disorders were labeled recessive and (rarely) dominant, like autosomal disorders. However, geneticists now recognize that differences in how the X chromosome is inherited, as well as biological processes specific to the X chromosome, make those labels inaccurate. Note that no PI-causing variants have been found in genes on the Y chromosome, so Y-linked inheritance is not covered here.
No matter how a PI is inherited, the chances of a child having PI, being a carrier, or neither remain the same in all pregnancies between the same biological mother and father. Like rolling dice, the outcome of each pregnancy is independent and is not affected by previous pregnancies.
Sometimes, variants aren’t there when a fertilized egg first begins growing but appear later in a baby’s development. These are called somatic variants—they are found in only some but not all of a person’s cells. Depending on which cells have the somatic variant, it can cause PI but may require special diagnostics and management to detect and treat. In many but not all cases, people with a somatic variant cannot pass down the variant to their children because the variant is not present in their sperm or egg cells.
It is important to understand that different variants in the same gene can cause different PIs. These different disorders may have different inheritance patterns, or the same inheritance pattern but a different way that they cause PI (mechanism). Why does this matter? These PIs may have overlapping symptoms, but immunologists may treat or manage them differently based on how the disorders are caused by each specific kind of variant.
In autosomal recessive disorders, the PI-causing variant has to be in both of a person’s copies of a particular gene to cause symptoms of the condition. Usually, each biological parent of the person has one copy of the PI-causing gene variant. The parents do not have the condition because their other, “backup” copy of the gene works well enough to “make up for” the copy with the variant. These unaffected parents are called carriers.
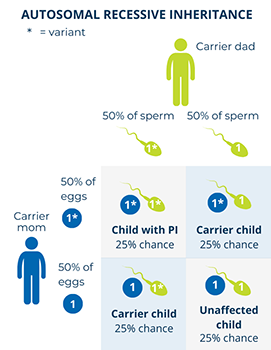
When both parents are carriers of an autosomal recessive PI-causing variant in a gene, there is:
- A 25% chance (1 in 4) that any child, regardless of sex, will be affected by the disorder.
- A 50% chance (1 in 2) that any child, regardless of sex, will inherit one copy of the variant and be an unaffected carrier for the disorder.
- A 25% chance (1 in 4) that the child, regardless of sex, will not inherit either variant, and will not be affected by the condition nor will they be a carrier.
Examples of autosomal recessive PIs include:
- Some forms of severe combined immunodeficiency (SCID).
- Adenosine deaminase (ADA) deficiency.
- Recombinase activating gene (RAG1/2) deficiency.
- JAK3 deficiency.
- Artemis SCID.
- MHC class II deficiency.
- Three forms of chronic granulomatous disease (CGD).
- Leukocyte adhesion deficiency (LAD) types 1-3.
- Most forms of primary hemophagocytic lymphohistiocytosis (HLH), including Chediak-Higashi syndrome.
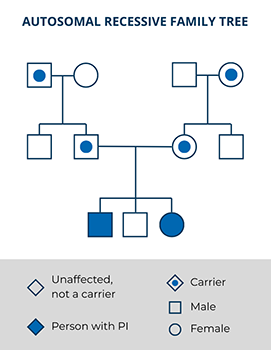
Autosomal recessive PIs are typically rare. Most people with an autosomal recessive PI do not have any other family members with the condition aside from, sometimes, their siblings. However, having a child with someone who is closely related to you by blood or who is from the same small community increases the chance that the child could have an autosomal recessive PI (e.g., Artemis SCID in Diné/Navajo populations). It’s important to be aware that genetic testing is not perfect and may not always be able to detect both PI-causing variants contributing to an autosomal recessive condition.
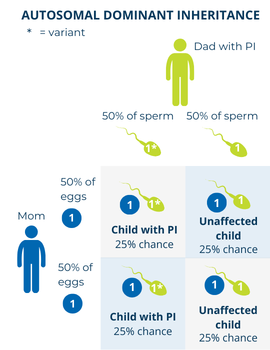
In autosomal dominant disorders, the PI-causing gene variant only has to be in one of a person’s two copies of a particular gene to cause symptoms of the condition. Because the variant is autosomal, both males and females can be affected. When someone with an autosomal dominant PI has a child, there is:
- A 50% chance (1 in 2) that any child, regardless of sex, will inherit the copy of the gene with the PI-causing variant and be affected.
- A 50% chance (1 in 2) that any child, regardless of sex, will inherit the copy of the gene without the PI-causing variant and not be affected.
It is important to note that if a person has an autosomal dominant condition, this does not mean that 50% or half of all of their children will inherit the condition. The 50% or 1 in 2 chance applies to each individual child, so it is possible for all or none of their children to inherit the condition.
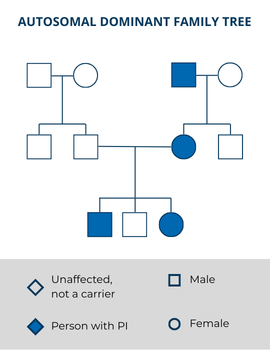
Sometimes, a person with an autosomal dominant condition is the first person with the disorder in their family. In this case, the PI-causing variant arose randomly in the egg or the sperm, or very early in the baby’s development—this is called a de novo mutation. Even though this person’s PI-causing variant was not inherited, they can still pass it on to their children.
Examples of autosomal dominant PIs include:
- Hyper IgE syndrome, due to STAT3 loss of function (LOF; Jobs syndrome).
- Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome.
- Some rare forms of IFN-γ/IL-12 pathway disorders.
- STAT1 and STAT3 gain-of-function (GOF).
- CTLA-4 haploinsufficiency.
- Activated PI3K-delta syndromes 1 and 2 (APDS1/2).
- Haploinsufficiency of A20 (HA20).
- Cryopyrin-associated periodic syndromes (CAPS) due to NLRP3 GOF.
If the gene variant that causes PI is on the X chromosome, the disorder is X-linked. Many, but not all, variants that cause X-linked PI can be offset by having a working “back-up” copy of the same gene—in other words, by having a second X chromosome without the variant. If a genetic male’s X chromosome has a PI-causing gene variant, they will have the disorder because they do not have a second “backup copy” of that gene (remember that the Y chromosome does not have the same set of genes as the X chromosome). Because of this, X-linked PIs are much more common in genetic males than genetic females. A family history may show many males related through their mothers who have the disorder.
Genetic females have two X chromosomes and, in many cases, must inherit two copies of the X-linked variant—one from each parent—to have an X-linked PI. Historically, genetic females who inherited one copy of an X-linked variant were considered carriers who did not have any symptoms, similar to carriers of autosomal recessive PIs. Healthcare providers assumed that their second, working copy of the X-linked gene made up for the copy with the variant.
However, this assumption is not entirely accurate. Females with one copy of a PI-causing X-linked variant can have symptoms, and often in different ways from affected males. This means they do not always have milder, later-onset forms of PI compared to males but may have different symptoms altogether.
One of the many ways females with one copy of an X-linked variant can have symptoms is through a natural biological process called X-inactivation. This process happens during fetal development when each of a genetic female’s cells shuts down (inactivates) one of their copies of the X chromosome. Which X chromosome each cell inactivates is random, so some cells inactivate the X chromosome with the PI-causing variant, and other cells inactivate the X chromosome without the variant. If the X chromosome that has the working copy of the gene is inactivated, then it cannot be the “backup” that makes up for the copy with the variant. Depending on which cells and how many cells have inactivated the X chromosome with the working copy of the gene, genetic females can also be affected by X-linked PI.
Similar to carriers of autosomal recessive PIs, carriers of X-linked PIs can pass on the gene variant to their children. Because each pregnancy outcome is independent, if a mother is a carrier, it is possible for all of her sons to have the X-linked disorder.
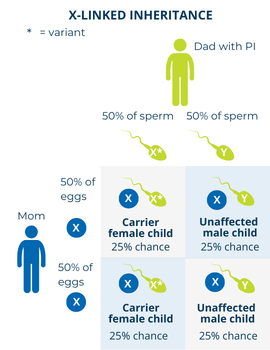
The chances of a child having an X-linked disorder or being a carrier depend on which parent has the PI-causing variant and the sex of the child.
If a father with an X-linked PI has children with someone who is not a carrier, there is:
- A 50% chance (1 in 2) of having a female child who is a carrier and may be affected or unaffected.
- A 50% chance (1 in 2) of having a male child who does not have the disorder and is not a carrier.
If a father with an X-linked PI has children with someone who is a carrier, the possibilities change. However, this situation is rare. There is:
- A 25% chance (1 in 4) of having a male child who has the disorder.
- A 25% chance (1 in 4) of having a male child who does not have the disorder.
- A 25% chance (1 in 4) of having a female child who has the disorder.
- A 25% chance (1 in 4) of having a female child who is a carrier and may be affected or unaffected.
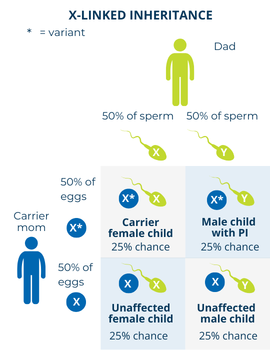
If a mother is a carrier of an X-linked PI-causing gene variant and has children with someone who does not have the disorder, there is:
- A 25% chance (1 in 4) of having a male child who has the disorder.
- A 25% chance (1 in 4) of having a male child who does not have the disorder.
- A 25% chance (1 in 4) of having a female child who is a carrier and may be affected or unaffected.
- A 25% chance (1 in 4) of having a female child who does not have the disorder and is not a carrier.
Variants in genes on the X chromosome can also happen spontaneously, or de novo. If the X-linked variant arises in the egg or very early in the baby’s development, male children will have the disorder while female children will be carriers. There will be no previous family history of the disorder. Even though this person’s PI-causing variant was not inherited, they can still pass it on to their children.
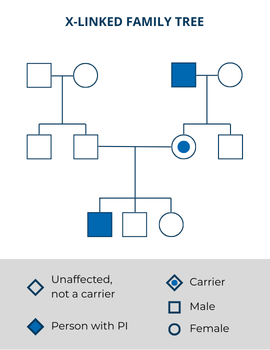
Examples of X-linked PIs include:
- X-linked agammaglobulinemia (XLA).
- Wiskott-Aldrich syndrome (WAS).
- SCID caused by common gamma chain variants (IL2RG deficiency).
- X-linked lymphoproliferative disease 1 (SAP deficiency) or 2 (XIAP deficiency).
- The most common form of chronic granulomatous disease (CGD).
- Properdin deficiency.
The cells in our body divide throughout our lifetime to help us grow and repair our tissues. There can sometimes be genetic changes that happen to our DNA during one of these divisions and lead to what we call somatic variants. In these cases, the person has a mixture of some cells with the gene variant and some cells without the gene variant.
Most of the time, when somatic variants happen, they do not cause a genetic disorder. This is because only some of the person’s cells have the gene variant. For example, a somatic variant in a gene that’s important for T cells won’t have any effect if the variant is only in a person’s liver cells. Also, if the gene variant is on an autosome, even the affected cells have another working copy of the gene.
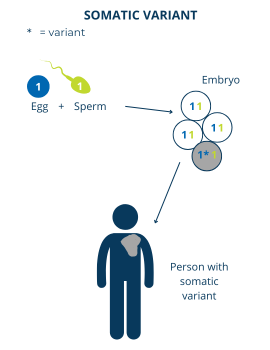
However, there are some cases where a somatic gene variant can cause PI. In these cases, the gene variant happens to be in cells where the function of that gene is important, even in small amounts.
Usually, people who have PI caused by somatic variants cannot pass the variant on to their children. However, the chance of this is not zero. It is possible that someone with a somatic variant can have some egg or sperm cells that contain the variant. If this happens, it will look like the child has a de novo variant, when in fact they inherited their variant.
Examples of PIs caused by somatic variants include:
- Late-onset cryopyrin-associated periodic syndromes (CAPS) due to NLRP3 GOF.
- RAS-associated leukoproliferative disorders.
- VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome) due to somatic UBA1 variants.
- Autoimmune lymphoproliferative syndrome (ALPS) due to somatic FAS variants.
- TLR8 gain-of-function (GOF).
Understanding PI more clearly

What is PI?
Also known as inborn errors of immunity (IEI), PIs are a group of > 550 rare, chronic conditions where a part of your immune system is missing or does not function correctly.
Learn the signs and symptoms

Your immune system and PI
To better understand PI, it's helpful to know more about how the immune system works.
Read about your immune system

Genetic testing
Genetic testing can be a critical step in getting the correct diagnosis.
How genetic testing works
This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice. Additionally, links to other resources and websites are shared for informational purposes only and should not be considered an endorsement by the Immune Deficiency Foundation.
Adapted from the IDF Patient & Family Handbook for Primary Immunodeficiency Diseases, Sixth Edition.
Copyright ©2019 by Immune Deficiency Foundation, USA.
Sign up for updates
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.
